
|
Photophysics and Photochemistry of Transition Metal Compounds |
| Home Research Members Collaborations Publications |



|
 |
|||||||
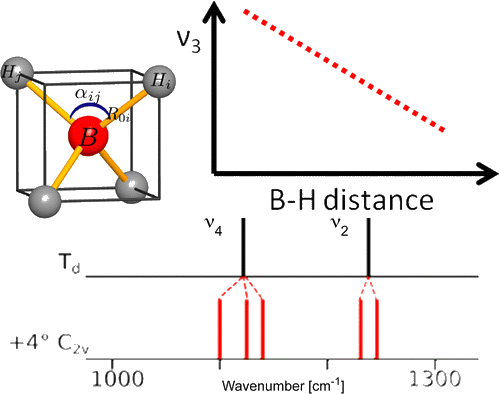
Among the different potential hydrogen storage materials, borohydrides have been largely investigated because of their high gravimetric and volumetric hydrogen content. In the analysis of borohydrides, vibrational spectroscopy plays an important role since it gives information on the local structure of the BH4â ion inside the solid. Here the GF method, developed by Wilson, is used in order to determine the local symmetry of BH4â in solid borohydrides starting from their vibrational spectra. Two different cases of deformations of BH4â are considered. In the first case, the effects of small angular variations on the vibrational spectra of borohydrides will be taken into account; starting from the splitting of the bands corresponding to the deformation modes, the angular deformations will be estimated. In the second one, the BH4â under chemical pressure (in different cubic alkali halides) is considered; in this case, the symmetry of the BH4â remains Td, while the bond lengths change according to the pressure experienced. Different practical examples will be illustrated. | ||||||||
 |
|
|||||||
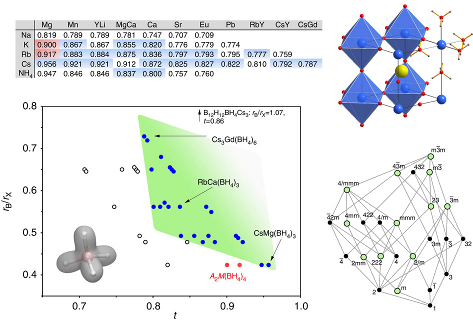
Perovskite materials host an incredible variety of functionalities. Although the lightest element, hydrogen, is rarely encountered in oxide perovskite lattices, it was recently observed as the hydride anion Hâ, substituting for the oxide anion in âBaTiO3. Here we present a series of 30 new complex hydride perovskite-type materials, based on the non-spherical âtetrahydroborate anion âBH4â and new synthesis protocols involving rare-earth elements. Photophysical, electronic and âhydrogen storage properties are discussed, along with counterintuitive trends in structural behaviour. The electronic structure is investigated theoretically with density functional theory solid-state calculations. BH4-specific anion dynamics are introduced to perovskites, mediating mechanisms that freeze lattice instabilities and generate supercells of up to 16 Ã the unit cell volume in AB(BH4)3. In this view, homopolar hydridic di-hydrogen contacts arise as a potential tool with which to tailor crystal symmetries, thus merging concepts of molecular chemistry with ceramic-like host lattices. Furthermore, anion mixing âBH4ââXâ (Xâ=Clâ, Brâ, Iâ) provides a link to the known ABX3 halides. | ||||||||
|
 |
|||||||
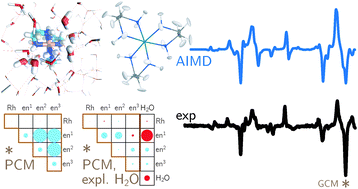
Backscattered Raman optical activity (ROA) spectra are measured for Î- and Î-tris-(ethylenediamine)rhodium(III) chloride in aqueous solution. In addition, the spectra of the four possible conformers in the Î configuration are investigated by ab initio calculations. The Î(δδδ) conformer is in best agreement with experimental spectra and examined in more details. The two most stable conformers according to the calculations are not compatible with the experimental ROA spectrum. Insights into the origin of observed band intensities are obtained by means of group coupling matrices. The influence of the first solvation shell is explored via an ab initio molecular dynamics simulation. Taking explicit solvent molecules into account further improves the agreement between calculation and experiment. Analysis of selected normal modes using group coupling matrices shows that solvent molecules lead to normal mode rotation and thus contribute to the ROA intensity, whereas the contribution of the Rh can be neglected. | ||||||||
 |
|
|||||||
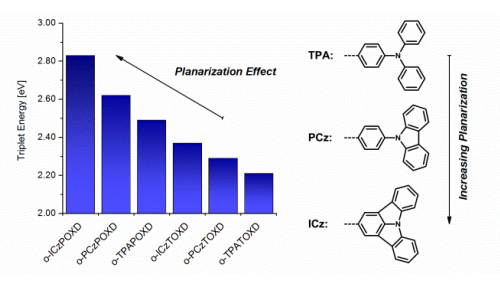
A series of 6 novel triarylamine-containing oxadiazole compounds (o-PCzPOXD, o-ICzPOXD, o-TPATOXD, o-PCzTOXD, o-ICzTOXD, o-CzTOXD) have been designed, synthesized and characterized concerning applications as host materials in PHOLED devices. To further improve the ortho-linkage concept, the impact of incorporating planarized electron-donating triarylamine (TAA) structures on intramolecular charge transfer was examined. The effect was evaluated for two series of electron-accepting oxadiazole scaffolds, realizing ortho-linkage on the benzene (POXD) and the thiophene (TOXD) core. Thermal analysis shows increased glass-transition temperatures for planarized structures indicating an improved morphological stability. A higher degree of planarization also results in significantly increased singlet and triplet energy values, revealing the impact on the intramolecular charge transfer. Employing the developed materials, red (o-TPATOXD: CEmax: 28.8 cd A-1, EQEmax: 16.9%), green (o-PCzPOXD: CEmax: 62.9 cd A-1, EQEmax: 17.1%) and blue (o-PCzPOXD: CEmax: 29.8 cd A-1, EQEmax: 13.4%) devices were achieved showing remarkably low efficiency roll-off for planarized donors. Hence, this is the first report of efficient blue devices for this specific class of host materials. It is proposed that the results correlate with an increasing ortho-linkage effect and decreasing donor strength of the TAA moiety by planarization and, thus, tackling one of the major challenges in PHOLED research: improving both triplet energy and compound stability. | ||||||||

|
 |
|||||||
A comparison of the vibrational spectra of many inorganic borohydrides allows us to distinguish compounds with isolated BH4- ions and compounds containing complex ions such as Sc(BH4)4-. The characteristic spectral features of both types of compounds are identified, showing that the BâH bonding is quite different in both cases. A detailed analysis of the vibrations of the isolated BH4-Â ions provides new information about their local structure. Angular deformations of individual borohydride ion are analyzed quantitatively. It appears that the compounds containing isolated BH4-Â ions belong to those with the most electropositive cations and the highest decomposition temperature, while the complex borohydrides show significantly lower decomposition temperatures and possible diborane formation. | ||||||||
 |
|
|||||||
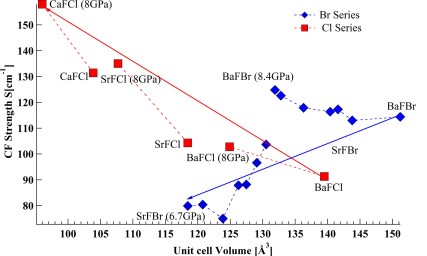
The emission spectra of Sm2+ doped in BaFBr and SrFBr hosts were measured at 10 K from ambient pressure to 8 GPa. The crystal field energy levels determined from the emission spectra were used to extract the free ion parameters (Fk and ζ ) and crystal field parameters (Bqk). The variation of Fk and ζ as a function of pressure was studied systematically and was discussed in relation to the central field and symmetry restricted covalency models. The change of the spin orbit coupling parameter (ζ) with pressure for SrFBr:Sm2+ showed very different behavior than in other matlockite hosts. Moreover the variation of Bqk under pressure was studied. The pressure dependence of the Bqk was described quantitatively using the Superposition Model (SM) with the help of structural parameters as a function of pressure, obtained from periodic DFT calculations. The validity of the SM was tested for Sm2+ in BaFBr and SrFBr. It is shown that this model does not apply to SrFBr, in contrast to other matlockite host materials. | ||||||||
|
 |
|||||||
Four novel bimetallic borohydrides have been discovered, K2M(BH4)4 (M = Mg or Mn), K3Mg(BH4)5, and KMn(BH4)3, and are carefully investigated structurally as well as regarding their decomposition reaction mechanism by means of in situ synchrotron radiation powder X-ray diffraction (SR-PXD), vibrational spectroscopies (Raman and IR), thermal analysis (TGA and DTA), and ab initio density functional theory (DFT) calculations. Mechano-chemical synthesis (ball-milling) using the reactants KBH4, α-Mg(BH4)2, and α-Mn(BH4)2 ensures chlorine-free reaction products. A detailed structural analysis reveals significant similarities as well as surprising differences among the two isomorphs K2M(BH4)4, most importantly concerning the extent to which the complex anion [M(BH4)4]2â is isolated in the structure. Anisotropic thermal expansion and an increase in symmetry at high temperatures in K3Mg(BH4)5 is ascribed to the motion of BH4 groups inducing hydrogen repulsive effects, and the dynamics of K3Mg(BH4)5 are investigated. Decomposition in the manganese system proceeds via the formation of KMn(BH4)3, the first perovkite type borohydride reported to date. | ||||||||
 |
|
|||||||
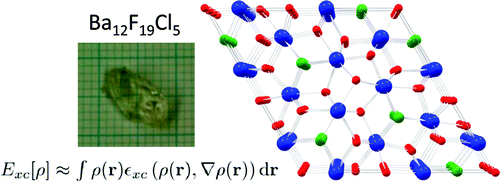
The crystal chemistry of the barium fluoride chloride system is studied both experimentally and theoretically. Different synthetic approaches yield nanocrystalline materials as well as large single crystals. The crystalline phases identified so far are BaFCl, Ba12F19Cl5 and Ba7F12Cl2 (in two modifications) and compared with analogous compounds. It is demonstrated that the compound Ba2F3Cl reported by Fessenden and Lewin 50 years ago corresponds to Ba7F12Cl2. The phase diagram of the BaCl2 â BaF2 system is reinvestigated for fluoride mole fractions between 0.5 and 1. The peritectic formation of Ba12F19Cl5 is observed. Periodic DFT calculations are performed for all structures in this system, including a hypothetical structure for Ba2F3Cl, based on the experimental structure of Ba2H3Cl. The energy of formation of the different barium fluoride chloride compounds from BaCl2 and BaF2 (normalized for one barium atom per formula unit), as calculated by DFT at 0K, is within only about ± 15 kJ/mol. Comparison with recent experimental results on calcium and strontium hydride chloride (bromide) compounds, suggest the possibility of a mutual exclusion between the M2X3Y and M7X12Y2 (M = Ca, Sr, Ba, Pb, X = H, F, Y = Cl,Br) structures. The single crystal structure of PbFBr is also reported. | ||||||||
|
||||||||
The structural and vibrational properties of the isostructural compounds Ca2FeH6Â and Sr2RuH6Â are determined by periodic DFT calculations and compared with their previously published experimental crystal structures as well as new experimental vibrational data. The analysis of the vibrational data is extended to the whole series of alkaline-earth iron and ruthenium hydrides A2TH6Â (A = Mg,Ca,Sr; T = Fe, Ru) in order to identify correlations between selected frequencies and the T-H bond length. The bulk moduli of Ca2FeH6Â and Sr2RuH6Â have also been determined within DFT. Their calculated values prove to compare well with the experimental values reported for Mg2FeH6Â and several other compounds of this structure. | ||||||||
|
 |
|||||||
The new double-cation Al-Li-borohydride is an attractive candidate material for hydrogen storage due to a very low hydrogen desorption temperature (~70 °C) combined with a high hydrogen density (17.2 wt %). It was synthesised by high-energy ball milling of AlCl3 and LiBH4. The structure of the compound was determined from image-plate synchrotron powder diffraction supported by DFT calculations. The material shows a unique 3D framework structure within the borohydrides (space group=P-43n, a=11.3640(3) Ã
). The unexpected composition Al3Li4(BH4)13 can be rationalized on the basis of a complex cation [(BH4)Li4]3+ and a complex anion [Al(BH4)4]-. The refinements from synchrotron powder diffraction of different samples revealed the presence of limited amounts of chloride ions replacing the borohydride on one site. In situ Raman spectroscopy, differential scanning calorimetry (DSC), thermogravimetry (TG) and thermal desorption measurements were used to study the decomposition pathway of the compound. Al-Li-borohydride decomposes at ~70 °C, forming LiBH4. The high mass loss of about 20 % during the decomposition indicates the release of not only hydrogen but also diborane. | ||||||||
 |
|
|||||||
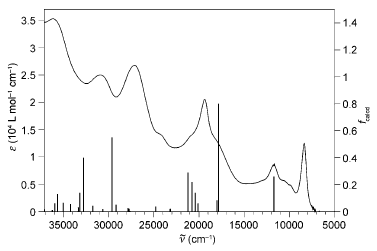
Electronic absorption spectrum of 1 in DMF solution at room temperature, together with the calculated oscillator strengths. | ||||||||
|
||||||||
The influence of pressure on the structural and vibrational properties of a2RuH6has been investigated using periodic density functional theory calculations performed at the local density approximation (LDA) and generalized gradient approximation (GGA) levels. At ambient pressure, the calculated structure and vibrational frequencies are in satisfactory agreement with experimental data. The calculated em>P-Vcurves could be fitted with the Vinet equation of state, yielding em>B0=67.6and em>B0=58.5  GPaat the LDA and GGA levels, respectively, and em>B0â²=4.0at both theoretical levels. The unit cell parameter is found to decrease faster with increasing pressure than the RuâH bond length. The calculated pressure dependence of the vibrational frequencies agrees well with experiment for em>ν5(T2g)but not for em>ν9(A1g) | ||||||||
Download this list in format RIS
 EndNote
EndNote  BibTex
BibTex  PDF XML
PDF XML Last update Friday December 08 2017